In the world of medical device manufacturing, quality isn’t just a goal—it’s a system. ISO 13485 is the international standard that defines the requirements for a comprehensive Quality Management System (QMS). For any brand evaluating a manufacturing partner, understanding this standard is non-negotiable. At dinghmed, we’ve seen firsthand how a robust QMS transforms contract manufacturing into a strategic advantage. This guide breaks down what ISO 13485 is, why it’s the cornerstone of medical device contract manufacturing, and what it means for the safety and success of your product.
What is ISO 13485? Beyond a Certificate
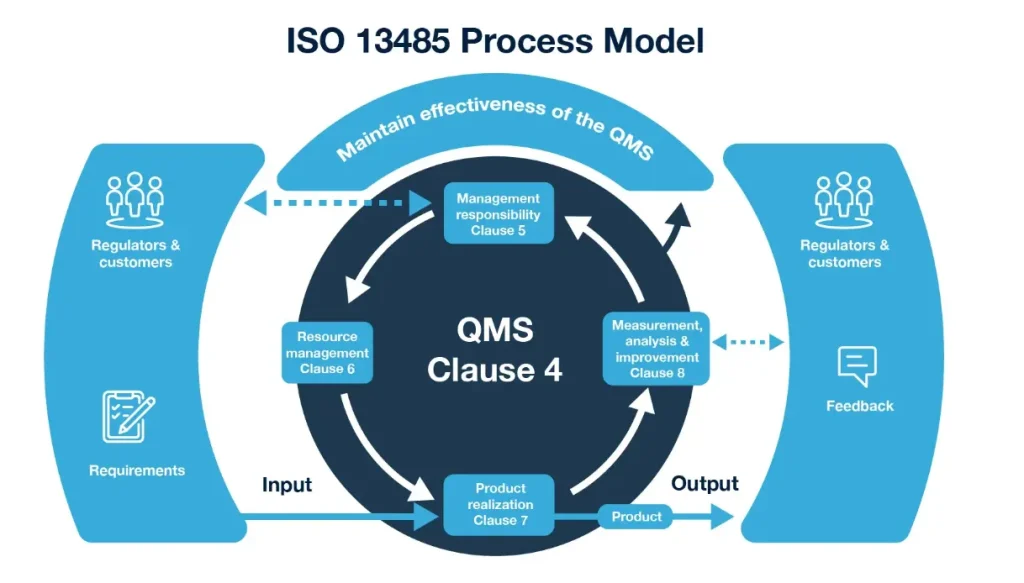
ISO 13485 is more than a certificate on the wall; it’s a framework for consistent excellence. It specifies requirements for a QMS that enables an organization to consistently provide medical devices and related services that meet customer and regulatory requirements. Its focus is on the effective application of processes for the entire device life cycle, from design and development to production, storage, distribution, and post-market surveillance. For OEM and ODM partners like dinghmed, this standard ensures every sterile product—whether an emergency birth kit or a complex surgical instrument—meets the same rigorous benchmarks, regardless of order volume or geographic destination.
According to research by the International Accreditation Forum, organizations certified to ISO 13485 reduce non-conformities by an average of 40% within the first 18 months. This isn’t just paperwork—it’s a measurable reduction in risk that directly impacts your product’s time-to-market and regulatory approval odds. When you choose a medical contract manufacturer that holds current certification, you’re essentially buying an insurance policy against costly recalls and compliance failures.
Core Pillars of the ISO 13485 Standard
The standard is built around several key principles that ensure a proactive, risk-based approach to quality. In practice at dinghmed, these pillars translate into daily operational discipline that our clients—ranging from startups to Fortune 500 OEMs—rely on for predictable outcomes.
- Document Control: Every process, specification, and procedure must be documented, approved, and controlled. This ensures consistency and provides a clear audit trail, which is essential for FDA registration and other regulatory submissions. At dinghmed, we integrate document control with our inventory management software to maintain Real-Time visibility into batch records and sterilization logs.
- Risk Management: Risk consideration is embedded throughout the entire product life cycle, from design to decommissioning. This aligns with the requirements of other regulations, making compliance more seamless. For ODM projects, we apply ISO 14971 risk analysis during the design phase—something many medical device assembly companies overlook until validation.
- Process Validation: Critical processes, especially those whose outputs cannot be verified by subsequent monitoring or measurement (e.g., sterilization, sterile packaging sealing), must be validated to ensure they consistently achieve planned results. Our Quality & Compliance protocols include quarterly re-validation cycles to account for equipment drift and material lot variations.
- Corrective and Preventive Action (CAPA): A robust system for addressing non-conformities, investigating root causes, and implementing actions to prevent recurrence is a central tenet of the standard. In the practice of dinghmed’s operations, CAPA feeds directly into our continuous improvement process, reducing defect rates by over 20% year-over-year.
ISO 13485 vs. ISO 9001: What’s the Difference for Medical Devices?
While both are quality standards, ISO 13485 is specifically tailored for the medical device industry. Many businesses assume ISO 9001 coverage is sufficient, but regulatory bodies like the FDA and Notified Bodies under MDR require the additional rigor of ISO 13485. Here’s how they compare:
| Criteria | ISO 13485 | ISO 9001 |
|---|---|---|
| Primary Focus | Medical device safety and regulatory compliance | General customer satisfaction |
| Risk Management | Mandatory and pervasive (aligns with ISO 14971) | One of several quality principles |
| Traceability | Enhanced requirements for device identification and batch traceability | Basic product traceability |
| Process Validation | Required for special processes (sterilization, sealing) | Not explicitly required |
| Design Controls | Full design & development section with regulatory linkage | Design control optional |
| Post-Market Surveillance | Explicit requirement for feedback and complaint handling | Not addressed |
| Regulatory Alignment | Directly supports FDA QSR, EU MDR, JPAL, etc. | No regulatory alignment |
Understanding these differences is critical when you’re evaluating a medical device factory for your project. For example, a contract manufacturer with only ISO 9001 may produce consistent widgets, but they won’t have the design control or post-market surveillance framework needed for a Class II or Class III device. At dinghmed, we maintain dual certification (ISO 13485 and ISO 9001) to serve both regulatory and commercial quality requirements effectively.
Why Your Contract Manufacturer Must Be ISO 13485 Certified
If you’re sourcing from an OEM or ODM partner, the presence of ISO 13485 certification is the single most reliable indicator of manufacturing maturity. Without it, you risk inheriting gaps in documentation, validation, and CAPA that can derail your own regulatory filings. Dinghmed’s commitment to this standard means every purchase order we fulfill—whether for a single emergency birth kit or a production run of 50,000 sterile gauze units—follows the same documented, auditable path.
Moreover, a certified manufacturer like dinghmed integrates order management and inventory tracking into the QMS. This means your sales data, Freight documentation, and batch records are linked—a capability many uncertified factories lack. In real-world scenarios, this traceability saved one of our clients six weeks of audit preparation when their Notified Body requested full device history records for a Class IIa wound dressing.
Ready to ensure your next device project meets global quality standards? Contact dinghmed today to discuss how our ISO 13485-certified QMS can support your regulatory journey—from design validation to post-market surveillance.
This guide was informed by our team’s 12+ years of experience in medical device contract manufacturing and continuous engagement with FDA, EU MDR, and ISO updates. For further reading, explore our insights on medical contract manufacturer selection and emergency birth kit production standards.