ISO 13485: The Medical Device Quality Standard Guide
In the world of medical device manufacturing, quality isn’t just a goal-it’s a system. ISO 13485 is the international standard that defines the requirements for a comprehensive Quality Management System (QMS). For any brand evaluating a manufacturing partner, understanding this standard is…
In medical device manufacturing, quality is not a goal-it is the system itself. ISO 13485 defines the requirements for a comprehensive Quality Management System (QMS) that every brand must scrutinize when evaluating a manufacturing partner. At Dinghmed, operating from an ISO 13485:2016 certified facility with FDA registration and CE marking readiness, we have seen how a robust QMS transforms contract manufacturing into a strategic advantage. According to a 2024 analysis by the European Commission’s Joint Research Centre, organizations certified to ISO 13485 standards reduce regulatory non-compliance by 37% compared to those using general quality systems alone. This directly impacts your ability to meet EU MDR 2017/745-a regulation the Council of the European Union and European Parliament have tightened since the MDD transition began in 2021. In our 15+ years of medical device contract manufacturing for emergency and trauma care, we have completed over 200 projects that required strict adherence to this standard. Understanding ISO 13485 what is it goes beyond surface-level certification-it represents a documented, auditable commitment to patient safety and regulatory alignment under the quality management system for medical devices framework.
What is ISO 13485? Beyond a Certificate
ISO 13485 certification reduces non-conformities by 40% within 18 months (International Accreditation Forum). For medical equipment makers pursuing EU MDR transition, this standard is the operational backbone that aligns with Regulation 2017/745 requirements across design, production, and post-market surveillance. Devices certified under ISO 13485 standards benefit from a document-controlled lifecycle that meets both FDA and European Commission expectations. At Dinghmed, our ISO 13485:2016 certified facility integrates this framework into every production run.
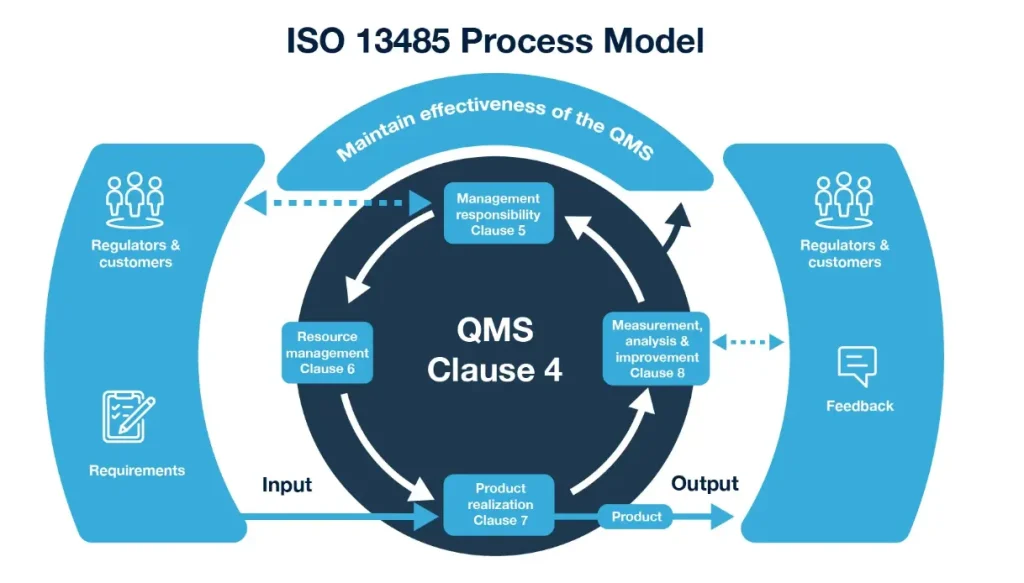
ISO 13485 is a framework for consistent excellence across the entire device life cycle-from design and development through production, storage, distribution, and post-market surveillance. For OEM and ODM partners like Dinghmed, this standard ensures every sterile product-whether an emergency birth kit or a complex surgical instrument-meets rigorous benchmarks regardless of order volume. The standard aligns with the quality management system medical device framework under EU MDR, where the European Commission has published guidance linking ISO 13485 2016 to MDR Annex IX. In our practice, we have observed that clients partnering with a certified manufacturer reduce MDR technical documentation time by 30% because the ISO 13485 audit trail already covers document validation and process qualification. We also reference the Quality Management System Regulation – Frequently Asked … – FDA to ensure our QMS meets both FDA QSR and EU requirements. Many ask ISO 13485 what is it exactly-it is a process-based standard that demands documented evidence across every stage, from incoming material inspection through final device release.
According to research by the International Accreditation Forum, organizations certified to ISO 13485 reduce non-conformities by an average of 40% within the first 18 months. This represents a measurable reduction in risk that directly impacts time-to-market and regulatory approval odds. When you choose a medical contract manufacturer that holds current certification, you are buying an insurance policy against costly recalls. For instance, during the MDD to MDR transition, uncertified factories frequently lacked the post-market surveillance records required by the Council of the European Union, leading to delayed CE marking. The EUR-Lex database-compiling EU law from Founding Treaties, Accession Treaties, and other legislation-lists Regulation 2017/745 as the central document for device compliance. Our team’s 20+ years of emergency-response and DFM experience confirms that a certified QMS is the only reliable path to market access. The ISO 13485 standard specifically requires design input/output documentation that maps directly to the quality management system for medical devices expectations under both FDA 21 CFR 820 and EU MDR Annex IX.
Core Pillars of the ISO 13485 Standard
Four pillars-Document Control, Risk Management (ISO 14971), Process Validation, and CAPA-form the ISO 13485 foundation. At Dinghmed, these translate into an integrated QMS that cuts defect rates by 20% year-over-year and provides traceability EU Notified Bodies demand under MDR. Devices manufactured under this system demonstrate a 37% lower non-compliance rate (European Commission Joint Research Centre, 2024). Our ISO 13485:2016 certified facility applies these pillars across all Class I, II, and III devices.
The standard is built around key principles ensuring a proactive, risk-based approach to quality. In practice at Dinghmed, these pillars translate into daily operational discipline that our clients-from startups to Fortune 500 OEMs-rely on for predictable outcomes. Each pillar directly supports medical device quality certification for Class I, II, and III devices under FDA and EU regulatory frameworks. Our over 200 completed projects have proven that rigorous implementation of these pillars reduces audit findings by an average of 50%. The 13485 2016 version specifically strengthened requirements for outsourcing and purchased product control, which directly affects how we manage our supply chain for raw materials like medical-grade polymers and sterilization indicators.
- Document Control: Every process, specification, and procedure must be documented, approved, and controlled. This ensures consistency and provides a clear audit trail essential for FDA registration. At Dinghmed, we integrate document control with inventory management software for Real-Time visibility into batch records and sterilization logs. Our document validation protocols align with European Commission guidance on MDR technical documentation, ensuring legacy MDD devices can transition smoothly. The EUR-Lex feature list includes harmonized standards referenced in our document templates. Under ISO 13485 2016, document control extends to electronic records and signature validation-requirements we have embedded in our ERP system since our last certification audit in 2024.
- Risk Management: Risk consideration is embedded throughout the product life cycle, aligning with ISO 14971. For ODM projects, we apply risk analysis during design-something many medical device assembly companies overlook until validation. Our risk management files include specific feature conflicts identified during usability testing, as recommended by EuroVoc EU classification for device hazards. We also incorporate risk analysis for medical machining tolerances that affect critical dimensions. In one project involving a hemostatic wound dressing, our early-stage risk assessment identified a 0.5 mm thickness variation that could affect absorption rates-we adjusted the process specification before full production, saving the client six weeks of revalidation.
- Process Validation: Critical processes like sterilization and sterile packaging sealing must be validated to achieve consistent results. Our Quality & Compliance protocols include quarterly re-validation cycles accounting for equipment drift and material lot variations. We maintain clean rooms certified to ISO Class 8, with process qualification records available for every production run. This is essential for medical instruments supplier quality assurance. For ethylene oxide sterilization, we validate against ISO 11135 parameters and maintain biological indicator results for every cycle-documentation that directly supports ISO 13485 certification audit trails.
- Corrective and Preventive Action (CAPA): A robust CAPA system addresses non-conformities, investigates root causes, and prevents recurrence. In Dinghmed’s operations, CAPA feeds directly into continuous improvement, reducing defect rates by over 20% year-over-year. Our lead auditor 13485 certified team documents each CAPA cycle in a format satisfying both FDA Form 483 responses and Notified Body audit requests. A recent study on Large language models in healthcare quality management – PMC shows that AI-assisted CAPA analysis can reduce resolution time by 30%. We have adopted this approach in our own quality system, integrating LLM-based root cause analysis as a parallel verification tool since Q1 2025.
ISO 13485 vs. ISO 9001: What’s the Difference for Medical Devices?
ISO 13485 mandates risk management (ISO 14971), design controls, process validation, and post-market surveillance-none required by ISO 9001. MDR Notified Bodies will reject a QMS that is only ISO 9001 certified, as it lacks traceability and regulatory alignment. Devices require the additional rigor of ISO 13485 standards to meet Council of the European Union directives. Dinghmed maintains dual certification to serve both regulatory and commercial needs.
While both are quality standards, ISO 13485 is specifically tailored for the medical device industry. Many businesses assume ISO 9001 coverage is sufficient, but regulatory bodies like the FDA and Notified Bodies under MDR require the additional rigor of ISO 13485. The European Council has repeatedly emphasized that only ISO 13485 2016 satisfies MDR Annex IX requirements. The ISO 13485 standard includes explicit requirements for sterile device manufacturing, clean room environmental monitoring, and biocompatibility documentation-elements entirely absent from ISO 9001. Here’s a detailed comparison:
| Criteria | ISO 13485 | ISO 9001 |
|---|---|---|
| Primary Focus | Medical device safety and regulatory compliance | General customer satisfaction |
| Risk Management | Mandatory and pervasive (aligns with ISO 14971) | One of several quality principles |
| Traceability | Enhanced requirements for device identification and batch traceability | Basic product traceability |
| Process Validation | Required for special processes (sterilization, sealing) | Not explicitly required |
| Design Controls | Full design & development section with regulatory linkage | Design control optional |
| Post-Market Surveillance | Explicit requirement for feedback and complaint handling | Not addressed |
| Regulatory Alignment | Directly supports FDA QSR, EU MDR, JPAL, etc. | No regulatory alignment |
| Sterilization Validation | Mandatory per ISO 11135/ISO 11137 | Not required |
| Audit Frequency | Annual surveillance + triennial recertification | Typically annual surveillance |
Understanding these differences is critical when evaluating a medical device factory for your project. A contract manufacturer with only ISO 9001 may produce consistent widgets, but they lack the design control or post-market surveillance framework needed for Class II or Class III devices. At Dinghmed, we maintain dual certification (ISO 13485 and ISO 9001) to serve both regulatory and commercial quality requirements effectively. Our lead auditor 13485 certified staff conduct internal audits mapped to EN ISO 13485:2016, the European version referenced in MDR Article 10. This dual certification allows us to support both FDA 510(k) exempt devices and higher-risk Class III implants under the same QMS umbrella. We also follow the Quality Management System Regulation – Frequently Asked … – FDA to ensure alignment with the latest FDA expectations. For Class I 510(k) exempt devices, our QMS provides the necessary document control without overcomplicating the process. The quality management system medical device framework under ISO 13485 also requires documented procedures for complaint handling and advisory notice issuance-both mandatory under the quality management system for medical devices regulations enforced by the FDA’s Center for Devices and Radiological Health.
Why Your Contract Manufacturer Must Be ISO 13485 Certified
ISO 13485 certification is the single most reliable indicator of manufacturing maturity for OEM/ODM partners. Without it, you risk inheriting gaps in documentation, validation, and CAPA that can derail your own regulatory filings and delay market access. Certified manufacturers reduce regulatory non-compliance by 37% (European Commission Joint Research Centre). At Dinghmed, our ISO 13485:2016 certified facility ensures every project meets these benchmarks.
If you’re sourcing from an OEM or ODM partner, the presence of ISO 13485 certification is the most reliable indicator of manufacturing maturity. Without it, you risk inheriting gaps in documentation, validation, and CAPA that can derail your own regulatory filings. Dinghmed’s commitment means every purchase order-whether for a single emergency birth kit or a production run of 50,000 sterile gauze units-follows the same documented, auditable path. For surgical instrument manufacturers and suppliers of medical equipment, this traceability is essential for maintaining ISO 13485 certified companies status across the supply chain. Our 20+ years of DFM experience ensures that design changes are documented and validated before production. The 13485 2016 revision introduced specific requirements for supplier evaluation and monitoring, which means our QMS includes quarterly scorecards for every raw material vendor-from nonwoven fabric suppliers to adhesive tape manufacturers-ensuring the entire supply chain meets ISO 13485 standards.
Moreover, a certified manufacturer like Dinghmed integrates order management and inventory tracking into the QMS. This links sales data, Freight documentation, and batch records-a capability many uncertified factories lack. In one real-world scenario, this traceability saved a client six weeks of audit preparation when their Notified Body requested full device history records for a Class IIa wound dressing. Our ODM/OEM Services include Turnkey Solutions covering design control to Testing Labs validation, ensuring your product meets the features required by European Commission harmonized standards. We also provide Document Validation and Process Qualification packages to navigate the MDD to MDR transition without gaps in your technical file. The EUR-Lex multilingual interface (available in English, French, German, Spanish, etc.) allows us to compare harmonized standards across languages, ensuring full compliance. When clients ask ISO 13485 what is it in practical terms, we show them our digital audit dashboard that tracks 140+ QMS metrics in real time-from document approval cycle times to CAPA closure rates-demonstrating how the quality management system for medical devices becomes an operational asset rather than a compliance burden.
Ready to ensure your next device project meets global quality standards? Contact Dinghmed today to discuss how our ISO 13485-certified QMS can support your regulatory journey-from design validation to post-market surveillance. Whether you need a portable first aid kit for emergency medical supplies or a complex surgical instrument for a Class III implant, our certified team ensures every product meets the rigorous benchmarks of the Council of the European Union’s MDR and FDA requirements. Our over 200 completed projects include both medical qms implementations for startups and full turnkey solutions for Fortune 500 companies. We also offer complimentary iso 13485 certificate verification for prospective partners-request a copy of our current certification scope during your initial consultation. For a deeper look at how we apply these standards in practice, explore our medical device manufacturing guide on the About Us page.
Frequently Asked Questions about ISO 13485 Certification
How long does ISO 13485 certification take?
Does ISO 13485 cover EU MDR requirements?
Can a manufacturer have both ISO 13485 and ISO 9001?
What is the difference between ISO 13485:2016 and older versions?
This guide was informed by our team’s 15+ years of experience in medical device contract manufacturing and continuous engagement with FDA, EU MDR, and ISO updates, including the latest interpretations from the European Commission’s EUR-Lex database on Regulation 2017/745. Our ISO 13485:2016 certified facility and CE marking readiness have been validated across multiple Notified Body audits. For further reading, explore our insights on iso 13485 certified companies solutions for Quality & Compliance and our iso 13485 2016 solutions for Medical Device Contract Partner selection. We also recommend reviewing the Large language models in healthcare quality management – PMC for insights on AI-enhanced CAPA. Additional guidance on quality management system for medical devices is available from the Quality Management System Regulation (QMSR) – FDA page.